Entry Point
Contents
Entry Point¶
All the command line tools is under the allcools command. The following chart illustrates their relationships.
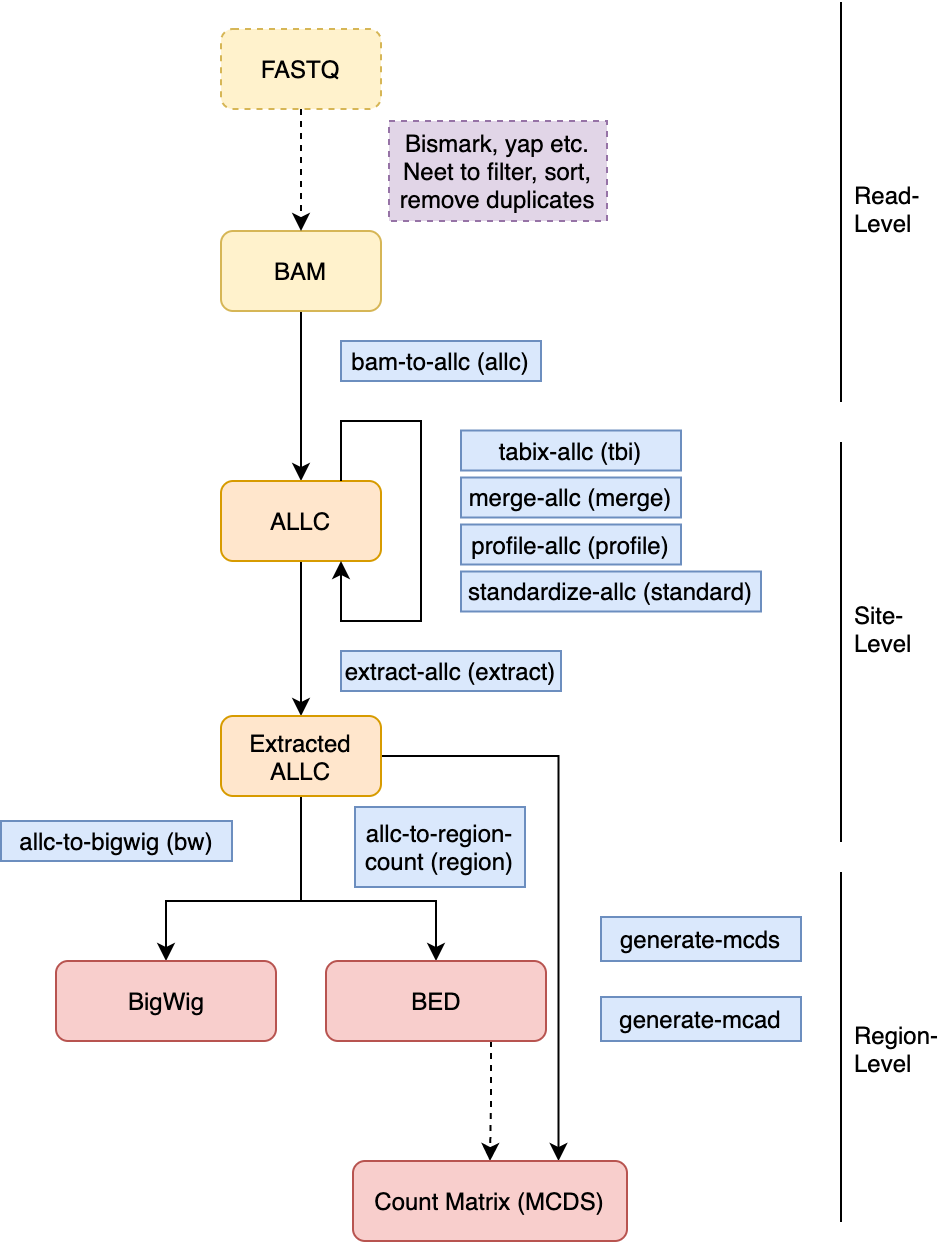
Fig. 8 ALLCools command line tools.¶
Usage¶
$ allcools -h
usage: allcools [-h] ...
The ALLCools command line toolkit contains multiple functions to manipulate the ALLC format,
a core file format that stores single base level methylation information.
Throughout this toolkit, we use bgzip/tabix to compress and index the ALLC file to allow
flexible data query from the ALLC file.
Current Tool List in ALLCools:
[Generate ALLC]
bam-to-allc - Generate 1 ALLC file from 1 position sorted BAM file via
samtools mpileup.
[Manipulate ALLC]
standardize-allc - Validate 1 ALLC file format, standardize the chromosome names,
compression format (bgzip) and index (tabix).
tabix-allc - A simple wrapper of tabix command to index 1 ALLC file.
profile-allc - Generate some summary statistics of 1 ALLC
merge-allc - Merge N ALLC files into 1 ALLC file
extract-allc - Extract information (strand, context) from 1 ALLC file
[Get Region Level]
allc-to-bigwig - Generate coverage (cov) and ratio (mc/cov) bigwig track files
from 1 ALLC file
allc-to-region-count - Count region level mc, cov by genome bins or provided BED files.
generate-mcds - Generate methylation dataset (MCDS) for a group of ALLC file and
different region sets. This is a convenient wrapper function for
a bunch of allc-to-region-count and xarray integration codes.
MCDS is inherit from xarray.DataSet
generate-mcad - Generate mCG hypo-methylation score AnnData dataset (MCAD) for
a group of ALLC file and one region set.
optional arguments:
-h, --help show this help message and exit
functions:
allc-motif-scan (motif)
Scan a list of ALLC files using a C-Motif
database.C-Motif Database, can be generated via
'allcools generate-cmotif-database' Save the
integrated multi-dimensional array into netCDF4 format
using xarray.
allc-to-bigwig (bw, 2bw)
Generate bigwig file(s) from 1 ALLC file.
allc-to-region-count (region, 2region)
Calculate mC and cov at regional level. Region can be
provided in 2 forms: 1. BED file, provided by
region_bed_paths, containing arbitrary regions and use
bedtools map to calculate; 2. Fix-size non-overlap
genome bins, provided by bin_sizes, Form 2 is much
faster to calculate than form 1. The output file is in
6-column bed-like format: chrom start end region_uid
mc cov
ame Motif enrichment analysis with AME from MEME Suite.
See AME doc for more information http://meme-
suite.org/doc/ame.html
bam-to-allc (allc, 2allc)
Take 1 position sorted BAM file, generate 1 ALLC file.
extract-allc (extract)
Extract information (strand, context) from 1 ALLC
file. Able to save to several different format.
generate-cmotif-database (cmotif-db)
Generate lookup table for motifs all the cytosines
belongs to. BED files are used to limit cytosine scan
in certain regions. Scanning motif over whole genome
is very noisy, better scan it in some functional part
of genome. The result files will be in the output
generate-mcad (mcad)
Generate MCAD from ALLC files and one region set.
generate-mcds (mcds)
Generate MCDS from ALLC files and region sets.
merge-allc (merge) Merge N ALLC files into 1 ALLC file
profile-allc (profile)
Generate some summary statistics of 1 ALLC.
standardize-allc (standard)
Standardize 1 ALLC file by checking: 1. No header in
the ALLC file; 2. Chromosome names in ALLC must be
exactly same as those in the chrom_size_path file; 3.
Output file will be bgzipped with .tbi index; 4.
Remove additional chromosome
(remove_additional_chrom=True) or raise KeyError if
unknown chromosome found (default)
tabix-allc (tbi) a simple wrapper of tabix command to index 1 ALLC file
Author: Hanqing Liu
See ALLCools documentation here: https://lhqing.github.io/ALLCools/intro.html