Feature Basic Filtering
Contents
Feature Basic Filtering¶
Purpose¶
Apply basic filters to remove these problematic features:
Extremly low coverage or high coverage features
ENCODE Blcaklist
Some chromosomes (usually, chrY and chrM)
Input¶
Cell metadata (after basic cell filter)
MCDS files
Output¶
FeatureList.BasicFilter.txt: List of feature ids passed all filters
Import¶
import pathlib
import pandas as pd
import seaborn as sns
from ALLCools.mcds import MCDS
sns.set_context(context='notebook', font_scale=1.3)
Parameters¶
# change this to the path to your filtered metadata
metadata_path = 'CellMetadata.PassQC.csv.gz'
# change this to the paths to your MCDS files
mcds_path = '../../../data/Brain/snmC-seq2/Liu2021Nature.mcds'
# Dimension name used to do clustering
obs_dim = 'cell' # observation
var_dim = 'chrom100k' # feature
min_cov = 500
max_cov = 3000
# change this to the path to ENCODE blacklist.
# The ENCODE blacklist can be download from https://github.com/Boyle-Lab/Blacklist/
black_list_path = '../../../data/genome/mm10-blacklist.v2.bed.gz'
f = 0.2
exclude_chromosome = ['chrM', 'chrY']
Load Data¶
Metadata¶
metadata = pd.read_csv(metadata_path, index_col=0)
total_cells = metadata.shape[0]
print(f'Metadata of {total_cells} cells')
Metadata of 16985 cells
metadata.head()
| AllcPath | mCCCFrac | mCGFrac | mCGFracAdj | mCHFrac | mCHFracAdj | FinalReads | InputReads | MappedReads | DissectionRegion | BamFilteringRate | MappingRate | Plate | Col384 | Row384 | FANSDate | Slice | Sample | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 10E_M_0 | /gale/raidix/rdx-4/mapping/10E/CEMBA190625-10E... | 0.008198 | 0.822633 | 0.821166 | 0.041640 | 0.033718 | 1626504.0 | 4407752 | 2892347.0 | 10E | 0.562347 | 0.656195 | CEMBA190625-10E-1 | 0 | 0 | 190625 | 10 | 10E_190625 |
| 10E_M_1 | /gale/raidix/rdx-4/mapping/10E/CEMBA190625-10E... | 0.006019 | 0.743035 | 0.741479 | 0.024127 | 0.018218 | 2009998.0 | 5524084 | 3657352.0 | 10E | 0.549577 | 0.662074 | CEMBA190625-10E-1 | 0 | 1 | 190625 | 10 | 10E_190625 |
| 10E_M_10 | /gale/raidix/rdx-4/mapping/10E/CEMBA190625-10E... | 0.006569 | 0.750172 | 0.748520 | 0.027665 | 0.021235 | 1383636.0 | 3455260 | 2172987.0 | 10E | 0.636744 | 0.628892 | CEMBA190625-10E-1 | 19 | 0 | 190625 | 10 | 10E_190625 |
| 10E_M_101 | /gale/raidix/rdx-4/mapping/10E/CEMBA190625-10E... | 0.006353 | 0.760898 | 0.759369 | 0.026547 | 0.020323 | 2474670.0 | 7245482 | 4778768.0 | 10E | 0.517847 | 0.659551 | CEMBA190625-10E-1 | 18 | 3 | 190625 | 10 | 10E_190625 |
| 10E_M_102 | /gale/raidix/rdx-4/mapping/10E/CEMBA190625-10E... | 0.005409 | 0.752980 | 0.751637 | 0.019497 | 0.014164 | 2430290.0 | 7004754 | 4609570.0 | 10E | 0.527227 | 0.658063 | CEMBA190625-10E-1 | 19 | 2 | 190625 | 10 | 10E_190625 |
MCDS¶
mcds = MCDS.open(mcds_path,
var_dim='chrom100k',
use_obs=metadata.index)
total_feature = mcds.get_index(var_dim).size
mcds
<xarray.MCDS>
Dimensions: (cell: 16985, chrom100k: 27269, count_type: 2, mc_type: 2)
Coordinates:
* cell (cell) <U10 '10E_M_207' '10E_M_338' ... '9J_M_2969'
* chrom100k (chrom100k) int64 0 1 2 3 4 ... 27265 27266 27267 27268
chrom100k_bin_end (chrom100k) int64 dask.array<chunksize=(27269,), meta=np.ndarray>
chrom100k_bin_start (chrom100k) int64 dask.array<chunksize=(27269,), meta=np.ndarray>
chrom100k_chrom (chrom100k) <U5 dask.array<chunksize=(27269,), meta=np.ndarray>
* count_type (count_type) <U3 'mc' 'cov'
* mc_type (mc_type) <U3 'CGN' 'CHN'
strand_type <U4 'both'
Data variables:
chrom100k_da (cell, chrom100k, mc_type, count_type) uint16 dask.array<chunksize=(3397, 2479, 2, 2), meta=np.ndarray>
Attributes:
obs_dim: cell
var_dim: chrom100kFilter Features¶
Filter by mean coverage¶
mcds.add_feature_cov_mean()
Feature chrom100k mean cov across cells added in MCDS.coords['chrom100k_cov_mean'].
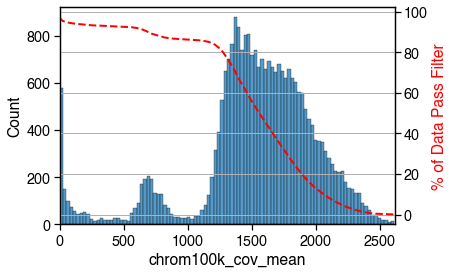
mcds = mcds.filter_feature_by_cov_mean(
min_cov=min_cov, # minimum coverage
max_cov=max_cov # Maximum coverage
)
Before cov mean filter: 27269 chrom100k
After cov mean filter: 25242 chrom100k 92.6%
Filter by ENCODE Blacklist¶
mcds = mcds.remove_black_list_region(
black_list_path=black_list_path,
f=f # Features having overlap > f with any black list region will be removed.
)
1189 chrom100k features removed due to overlapping (bedtools intersect -f 0.2) with black list regions.
Remove chromosomes¶
mcds = mcds.remove_chromosome(exclude_chromosome)
20 chrom100k features in ['chrM', 'chrY'] removed.
Save Feature List¶
print(
f'{mcds.get_index(var_dim).size} ({mcds.get_index(var_dim).size * 100 / total_feature:.1f}%) '
f'{var_dim} remained after all the basic filter.')
24045 (88.2%) chrom100k remained after all the basic filter.
with open('FeatureList.BasicFilter.txt', 'w') as f:
for var in mcds.get_index(var_dim).astype(str):
f.write(var + '\n')
mcds
<xarray.MCDS>
Dimensions: (cell: 16985, chrom100k: 24045, count_type: 2, mc_type: 2)
Coordinates:
* cell (cell) <U10 '10E_M_207' '10E_M_338' ... '9J_M_2969'
* chrom100k (chrom100k) int64 30 31 32 33 ... 26335 26336 26337
chrom100k_bin_end (chrom100k) int64 dask.array<chunksize=(24045,), meta=np.ndarray>
chrom100k_bin_start (chrom100k) int64 dask.array<chunksize=(24045,), meta=np.ndarray>
chrom100k_chrom (chrom100k) <U5 dask.array<chunksize=(24045,), meta=np.ndarray>
* count_type (count_type) <U3 'mc' 'cov'
* mc_type (mc_type) <U3 'CGN' 'CHN'
strand_type <U4 'both'
chrom100k_cov_mean (chrom100k) float64 1.4e+03 1.378e+03 ... 715.3 696.9
Data variables:
chrom100k_da (cell, chrom100k, mc_type, count_type) uint16 dask.array<chunksize=(3397, 2364, 2, 2), meta=np.ndarray>
Attributes:
obs_dim: cell
var_dim: chrom100k