DMR-Gene Correlation
Contents
DMR-Gene Correlation¶
Calculate the partial correlation between DMR and Genes using clusters/samples as features.
Import¶
import seaborn as sns
import matplotlib.pyplot as plt
from ALLCools.mcds import RegionDS
from ALLCools.mcds.correlation import region_correlation, get_corr_table
Load Data¶
# DMR data
dmr = RegionDS.open('../RegionDS/test_HIP/')
# gene data
gene = RegionDS.open('test_HIP_Cluster/', region_dim='geneslop2k')
Using dmr as region_dim
# global mC fractions, calculated from cell level MCDS by sum up cluster and genome base calls
sample_mch = gene.get_coords('sample_CHN')
sample_mcg = gene.get_coords('sample_CGN')
DMR Input¶
A 2-D sample-by-DMR DataArray, values are mCG fractions
data_a = dmr['dmr_da_frac'].transpose('sample', 'dmr')
data_a
<xarray.DataArray 'dmr_da_frac' (sample: 20, dmr: 131)>
array([[0.538462, 0.816129, 0.70068 , ..., 0.144186, 0.340136, 0.292444],
[0.717949, 0.97006 , 0.922078, ..., 0.03 , 0.142259, 0.249802],
[0.717949, 0.974227, 0.886179, ..., 0.011628, 0.085575, 0.196755],
...,
[0.888889, 0.670588, 0.038462, ..., 0.55 , 0.25974 , 0.621951],
[0.857143, 0.727823, 0.666667, ..., 0.295455, 0.562387, 0.544554],
[0.777778, 0.780822, 0.5625 , ..., 0.0625 , 0.297297, 0.292553]],
dtype=float32)
Coordinates:
* dmr (dmr) <U9 'chr1-0' 'chr1-1' 'chr1-2' ... 'chr19-121' 'chr19-122'
dmr_chrom (dmr) <U5 'chr1' 'chr1' 'chr1' ... 'chr19' 'chr19' 'chr19'
dmr_end (dmr) int64 10002172 10003542 10003967 ... 5099203 5099952
dmr_length (dmr) int64 2 305 54 2 2 2 10 ... 589 924 632 842 195 399 335
dmr_ndms (dmr) int64 1 7 2 1 1 1 2 13 3 2 1 ... 2 1 2 7 13 19 8 8 3 6 13
dmr_start (dmr) int64 10002170 10003237 10003913 ... 5098804 5099617
* sample (sample) <U18 'snm3C_ASC' 'snm3C_CA1' ... 'snmC_ODC' 'snmC_OPC'Gene Input¶
A 2-D sample-by-gene DataArray, values are cluster-normalized mCH fractions
data_b = gene.sel(
mc_type='CHN',
sample=data_a.get_index('sample'))['geneslop2k_da_frac'].transpose(
'sample', 'geneslop2k')
data_b
<xarray.DataArray 'geneslop2k_da_frac' (sample: 20, geneslop2k: 55487)>
[1109740 values with dtype=float64]
Coordinates:
mc_type <U3 'CHN'
* sample (sample) object 'snm3C_ASC' 'snm3C_CA1' ... 'snmC_OPC'
sample_CGN (sample) float64 0.7203 0.7387 0.7422 ... 0.7434 0.7196
sample_CHN (sample) float64 0.007835 0.02362 ... 0.01121 0.005743
strand_type <U4 'both'
* geneslop2k (geneslop2k) object 'ENSMUSG00000102693.1' ... 'ENSMUSG...
geneslop2k_chrom (geneslop2k) object 'chr1' 'chr1' 'chr1' ... 'chrM' 'chrM'
geneslop2k_end (geneslop2k) int64 3076321 3104124 3673497 ... 16299 16299
geneslop2k_start (geneslop2k) int64 3071252 3100015 3203900 ... 13288 13355Calculate correlations between DMR and genes¶
true_results, null_results = region_correlation(data_a,
data_b,
sample_mch,
sample_mcg,
method='pearson',
max_dist=1000000,
cpu=40,
null='sample',
null_n=100000)
Using dmr as region_dim
Using dmr_start and dmr_end to calculate pos
Using geneslop2k as region_dim
Using geneslop2k_start and geneslop2k_end to calculate pos
Calculating chr1
Calculating chr19
true_results.X.data.size, null_results.size
(19100, 19100)
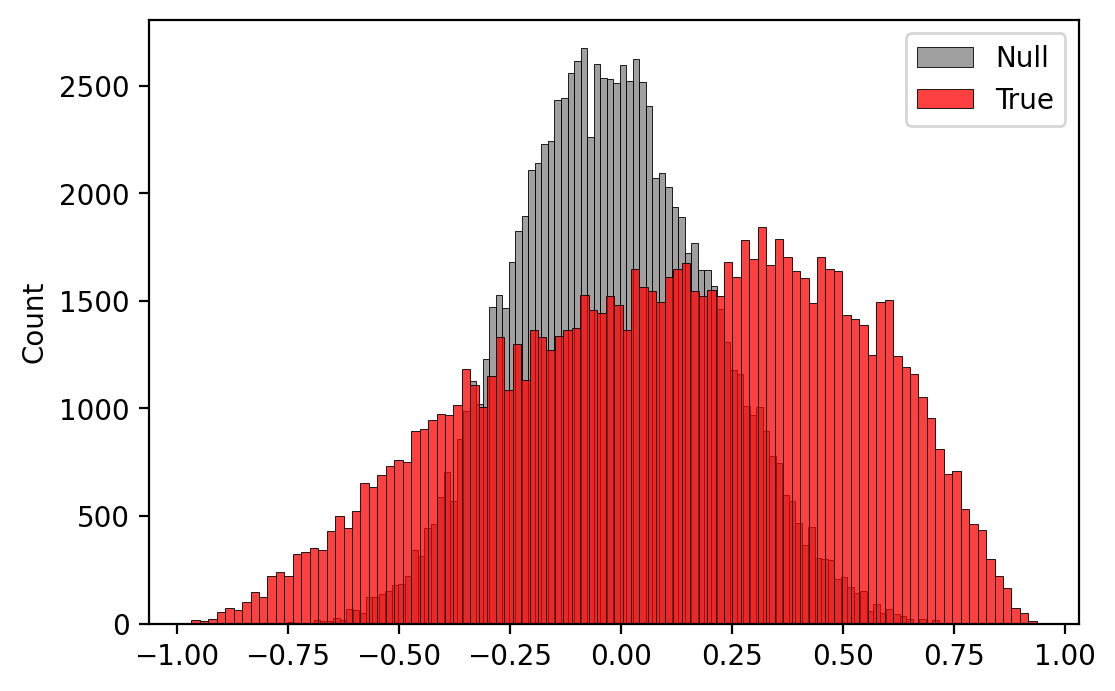
Plot correlation distribution¶
import numpy as np
fig, ax = plt.subplots(dpi=200)
sns.histplot(ax=ax,
data=np.random.choice(null_results, 100000),
label='Null',
bins=100,
color='grey')
sns.histplot(ax=ax,
data=np.random.choice(true_results.X.data, 100000),
label='True',
bins=100,
color='red')
ax.legend()
<matplotlib.legend.Legend at 0x7f5ce2c67b80>
findfont: Font family ['sans-serif'] not found. Falling back to DejaVu Sans.
findfont: Generic family 'sans-serif' not found because none of the following families were found: Helvetica
Get Significantly Correlated Pairs¶
corr_table = get_corr_table(true_results,
null_results,
region_dim_a=data_a.dims[1],
region_dim_b=data_b.dims[1],
direction='+',
alpha=0.03)
corr_table['pair_distance'] = corr_table['dmr_pos'] - corr_table['geneslop2k_pos']
corr_table
Correlation cutoff corr > 0.5499999999999998
2855 correlation pairs selected.
| dmr | geneslop2k | corr | dmr_pos | geneslop2k_pos | pair_distance | |
|---|---|---|---|---|---|---|
| 0 | chr1-0 | ENSMUSG00000025916.10 | 0.688226 | 10002171 | 9988879 | 13292 |
| 1 | chr1-2 | ENSMUSG00000102871.1 | 0.653428 | 10003940 | 9288550 | 715390 |
| 2 | chr1-3 | ENSMUSG00000067851.11 | 0.602079 | 10005478 | 10185119 | -179641 |
| 3 | chr1-3 | ENSMUSG00000098234.7 | 0.560595 | 10005478 | 9943037 | 62441 |
| 4 | chr1-3 | ENSMUSG00000045210.8 | 0.701606 | 10005478 | 9733501 | 271977 |
| ... | ... | ... | ... | ... | ... | ... |
| 2850 | chr19-122 | ENSMUSG00000024835.15 | 0.678979 | 5099784 | 4151326 | 948458 |
| 2851 | chr19-122 | ENSMUSG00000044724.9 | 0.811770 | 5099784 | 4142762 | 957022 |
| 2852 | chr19-122 | ENSMUSG00000024842.8 | 0.749665 | 5099784 | 4139727 | 960057 |
| 2853 | chr19-122 | ENSMUSG00000024845.17 | 0.655735 | 5099784 | 4129119 | 970665 |
| 2854 | chr19-122 | ENSMUSG00000024851.14 | 0.652242 | 5099784 | 4106980 | 992804 |
2855 rows × 6 columns
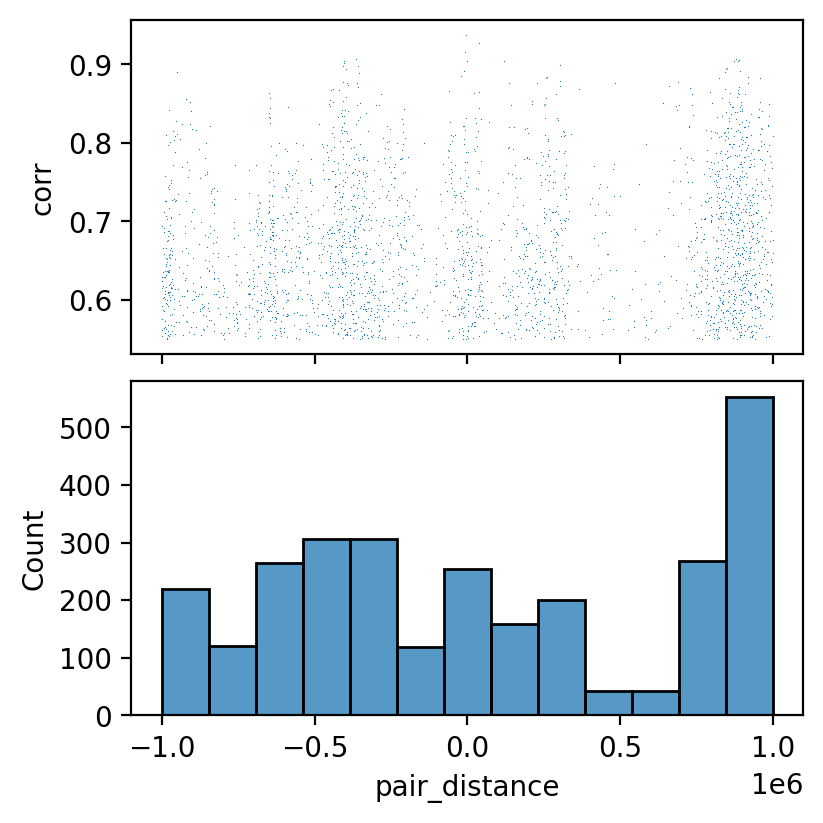
Plot distance and correlation distribution¶
fig, axes = plt.subplots(figsize=(4, 4), nrows=2, dpi=200, constrained_layout=True, sharex=True)
ax = axes[0]
sns.scatterplot(ax=ax,
data=corr_table,
x='pair_distance',
y='corr',
s=0.2,
linewidth=0)
ax = axes[1]
sns.histplot(corr_table['pair_distance'], ax=ax)
<AxesSubplot:xlabel='pair_distance', ylabel='Count'>